

Glycogen storage Disorders are biochemical disorders characterised by abnormal glycogen metabolism which can be presented by two major organs i.e, Liver and Muscle. In this particular section, we will provide a concise note on types of liver glycogen storage disorders aka Liver Glycogenosis.
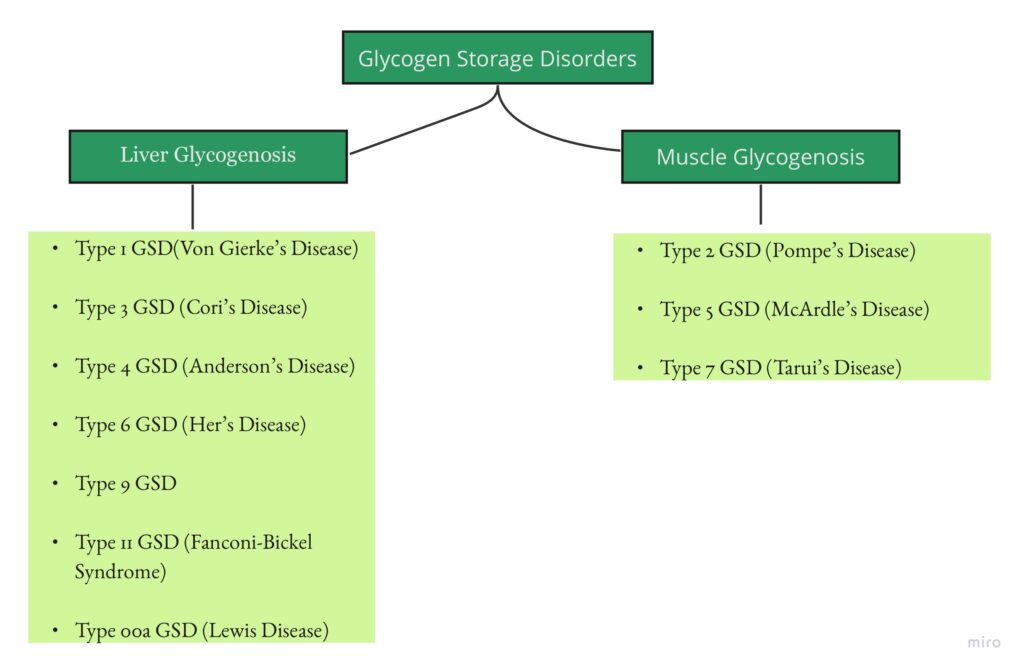
LIVER GLYCOGENOSIS :
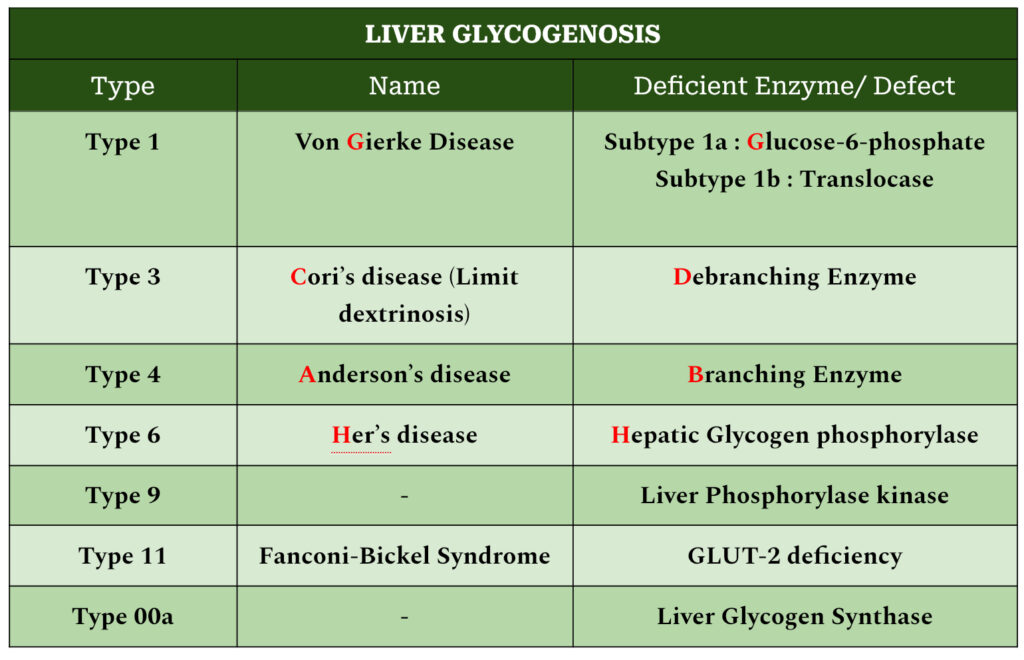
TYPE 1 GSD : VON GIERKE’S DISEASE
INHERITANCE : Autosomal recessive
DEFICIENT ENZYME :
In subtype 1a, Glucose-6-phosphatase
In subtype 1b, Translocase (carries glucose-6-phosphate across microsomal membrane)
ORGANS AFFECTED : Liver, Kidney, Intestinal Mucosa
CLINICAL FINDINGS :
- Neonatal period: Hypoglycemia and Lactic Acidosis
- 3-4 months: Hepatomegaly
- Physical features: Doll-like facies, fat cheeks, thin extremities, short stature, protuberant abdomen
- Growth retardation
- Easy bruising and epistaxis
- Diarrhoea and malnutrition (long-standing cases of subtype 1b)
LABORATORY FINDINGS :
- Prolonged bleeding time
- Hyperuricemia
- Elevation of TGs, LDL, Total cholesterol
- In type 1b, neutropenia (leading to recurrent bacterial infections and mucosal ulcerations)
LONG TERM COMPLICATIONS :
- Gout (presented at puberty)
- Polycystic ovaries and menorrhagia in women
- Pancreatitis
- Systemic hypertension
- Osteopenia/osteoporosis (seen in adults)
- Hepatic adenomas (2nd-3rd decade of life)
- End stage renal disease in patients who developed proteinuria, nephrocalcinosis and alteration in creatinine clearance
DIAGNOSIS :
- Clinical presentation
- Lab findings (mentioned above)
- Genetic testing
- Liver biopsy (historic value)- Distended by glycogen and fat, with large lipid vacuoles
TREATMENT :
- First line: Avoid fasting and frequent feedings
- Diet preferred: Complex carbohydrates with uncooked corn-starch
- For nephrocalcinosis: Citrate supplementation
- For hyperuricemia: Allopurinol
- For lipid abnormalities: HMG-CoA reductase inhibitors and fibrates
- For microalbuminuria: ACE inhibitors
- For liver adenoma: Surgical resection, radiofrequency ablation, percutaneous ethanol injection
- Liver and Kidney transplantation may be indicated
- For subtype 1b: G-CSF and Empagliflozin
TYPE 3 GSD : CORI’S DISEASE
INHERITANCE : Autosomal recessive
ENZYME INVOLVED : Glycogen debranching enzyme
CLASSIFICATION :
3a: Liver, skeletal muscle and cardiac involvement
3b: Primarily liver involvement
CLINICAL FINDINGS :
- Infancy: Hypoglycemia(ketotic or non-ketotic in only 50% of patients) , hepatomegaly (reduces with age), hyperlipidemia, short stature
- 3a: Skeletal myopathy and cardiomyopathy may be present with the above findings
- Liver: Hepatomegaly reduces with age but many patients in late adulthood have fibrosis, cirrhosis, liver failure, hepatocellular carcinoma, hepatic adenomas( less common than in Von Gierke’s disease)
- Heart: Left ventricular hypertrophy, arrhythmia
- Skeletomuscular: Muscle weakness (severe by 4th decade), exercise intolerance, osteoporosis
- Nervous system: Peripheral Neuropathy
- Polycystic ovaries in females
LABORATORY FINDINGS :
- Elevated ALT and AST
- Normal blood lactate and uric acid
- Elevation of TGs, LDL, Total cholesterol
- Serum creatine kinase may be elevated
DIAGNOSIS :
- Clinical presentation
- Lab findings (mentioned above)
- Genetic testing- DNA based analyses
- Liver biopsy (historic value)- distended hepatocytes, periportal fibrosis along with few fat infiltration
TREATMENT :
- Diet preferred: As gluconeogenesis is intact, high-protein diet and complex carbohydrates with uncooked corn-starch
- Dietary lipid modifications: High fat diet/ketogenic diet/supplementation of medium-chain TGs
- For lipid abnormalities: HMG-CoA reductase inhibitors and fibrates
- For liver adenoma: Surgical resection, radiofrequency ablation, percutaneous ethanol injection
- Liver and heart transplantation may be indicated
TYPE 4 GSD : ANDERSON’S DISEASE
ENZYME DEFICIENT : Branching Enzymes
CLASSIFICATION WITH FEATURES :
- Hepatic form- Failure to thrive, hypotonia, hepatomegaly, cirrhosis, failure (Death by 5 years), hypoglycemia (late finding secondary to liver lesions); small subset has extrahepatic involvement
- Neuromuscular forms-
It has 4 subtypes:
- Perinatal– Death in neonatal period
- Congenital– Death in neonatal period
- Childhood– Myopathy, cardiomyopathy, systemic findings
- Adult Polyglucosan Body Disease (APBD)– B/L Lower limb weakness, spasticity; neurogenic bladder; peripheral neuropathy; cognitive impairment
DEFINITIVE DIAGNOSIS :
- Demonstrate pathogenic variants in the GBE1 gene; or
- Enzyme deficiency in liver, muscle, cultured skin fibroblasts, leukocytes
TREATMENT :
- Liver transplantation
- Symptomatic for gait abnormalities and bladder dysfunction in APBD
TYPE 9 GSD
ENZYME INVOLVED : Liver Phosphorylase Kinase (PhK)
CLASSIFICATION :
- 9 alpha 2 (PHKA2 pathogenic variants):
- X-linked
- commonly seen
- high phenotypic variability
- Enzymes in liver, erythrocyte, leukocyte affected, not muscles
- 9 beta (PHKB pathogenic variants)
- 9 gamma 2 (PHKG2 pathogenic variants): Severe with early cirrhosis and fibrosis
- 9 alpha 1 (PRKAG2 pathogenic variants): Affect only muscles
- 9 gamma 1 (PRKAG2 pathogenic variants): Affect only muscles
TYPE 10 GSD : FANCONI-BICKEL SYNDROME
ENZYMES DEFICIENT : GLUT2 deficiency
FEATURES : Proximal renal tubular dysfunction leading to increased renal clearance of glucose, amino acids, phosphate and uric acid
Medicine is a science of uncertainty and an art of probability.
William osler
REFERENCE :
Harrison’s Principles of Internal Medicine, 21e Loscalzo J, Fauci A, Kasper D, Hauser S, Longo D, Jameson J. Loscalzo J, & Fauci A, & Kasper D, & Hauser S, & Longo D, & Jameson J(Eds.),Eds. Joseph Loscalzo, et al.